library(mrmhub)
mrmhub_enable_cli_color()
# Used only to compose the example chromatograms below.
library(ggplot2)
library(patchwork)Dataset 4 — Quantification & QC Report
Steroid Assay
1 Overview
This is an MRMhub postprocessing report for Dataset 4, a fully quantitative 15-analyte serum steroid panel measured with external calibration. The data are from an LC-MRM-MS analysis using the Steroid Panel LC-MS kit (IBL International, Germany), following the Instructions for Use except that LC-MS was performed on an Agilent 6495A triple quadrupole instrument. Only the Gradient 1 analysis was used, covering 15 analytes with their internal standards (66 MRM transitions monitored, including qualifiers, from which 64 features were integrated).
The dataset comprises system-suitability samples (SST, containing pure analyte and internal standards), blanks, calibrants (Cal A – Cal F), low and high quality controls (LQC, HQC) with assigned target concentrations, and, in place of study samples, 5 external quality assessment (EQA) samples (SKML (Jansen et al. 2014)).
These data were additionally processed end-to-end with a vendor tool (Agilent MassHunter Quantitative Analysis) and compared with MRMhub at both the peak-integration and the quantification stage, see MRMhub vs MassHunter.
ImportantData availability
The datasets and the full code used to generate this resource are deposited in the Zenodo record 10.5281/zenodo.15370293 (MRMhub-workflows).
Notemrmhub version
Rendered with mrmhub 0.9.9 (QUANT module of MRMhub, main branch): pak::pak("SLINGhub/MRMhub").
1.1 Setup
Load the package and call mrmhub_enable_cli_color() to render mrmhub’s console feedback in colour in the HTML output.
2 Raw Data Processing: Peak Picking and Integration
Peak picking and integration were performed with MRMhub-INTEGRATOR, as for Datasets 1 and 3. For a full description of the workflow and its parameters, see Workflow for Dataset 1 and the INTEGRATOR Manual.
2.1 Conversion of vendor raw files to mzML
INTEGRATOR requires the raw files in mzML format. The original Agilent .d files were converted using msconvert from the ProteoWizard software (https://proteowizard.sourceforge.io) with the following settings: output format = mzML; binary encoding precision = 32 bit; write index = true. See the MRMhub Manual for more details.
2.2 Preparing the INTEGRATOR input files
INTEGRATOR takes three input files: the global settings file (param.txt), the sample table listing the .mzML files in acquisition order, and the feature/transition table defining each transition by precursor and product m/z with its expected retention time. All input files used for this dataset are contained in MRMhub-Dataset4.zip in the Zenodo record above, for those who wish to re-run the integration. Of the 66 transitions monitored, 64 features were integrated: the 15 analytes, their 15 internal standards, and the qualifier transitions of both.
Two per-feature settings in the feature/transition table were adjusted for this panel. The peak_width parameter was set individually for several analytes, overriding the global value. The baseline mode was set to v_drop for most features, which places the horizontal baseline at the lowest intensity of the integrated region rather than at the default 5th percentile of the intensities in the integration window.
The batch was integrated in two subsets, exported as separate long-format tables: the ten replicate system-suitability injections (SST) and the 20 assay injections (ASSAY: three solvent blanks, one process blank, the calibrators Cal 0–Cal F, QC_Low1 and QC_High1, and seven injections of the five SKML EQA materials). This report reads the assay subset; the comparison with MassHunter uses both.
2.3 Running MRMhub-INTEGRATOR and Review of Results
The most recent release of INTEGRATOR was obtained from https://github.com/SLINGhub/MRMhub/releases. After preparing the input files, steps 1 to 4 of the application run consecutively to perform peak detection, peak picking, integration, and export of data and PDF results. The integration results are reported in ‘long.csv’, a long-format table with peak areas, apex retention times, peak widths and integration borders; here it was renamed to ‘Dataset4_MRMhub-INTEGRATOR_ASSAY.csv’. PDFs of the integrated transitions are written to the by_* folders.
The integration results were inspected for all features across all injections using the generated PDFs, and the settings above are the result of several rounds of parameter optimization with INTEGRATOR. Figure 1 shows six of these plots: three well-resolved QC peaks and, beneath them, three cases in which the analyte sits next to a co-eluting neighbour. The peak areas exported here are the input to the postprocessing that follows; they are compared against an independent Agilent MassHunter re-integration of the same raw files in the comparison workflow.
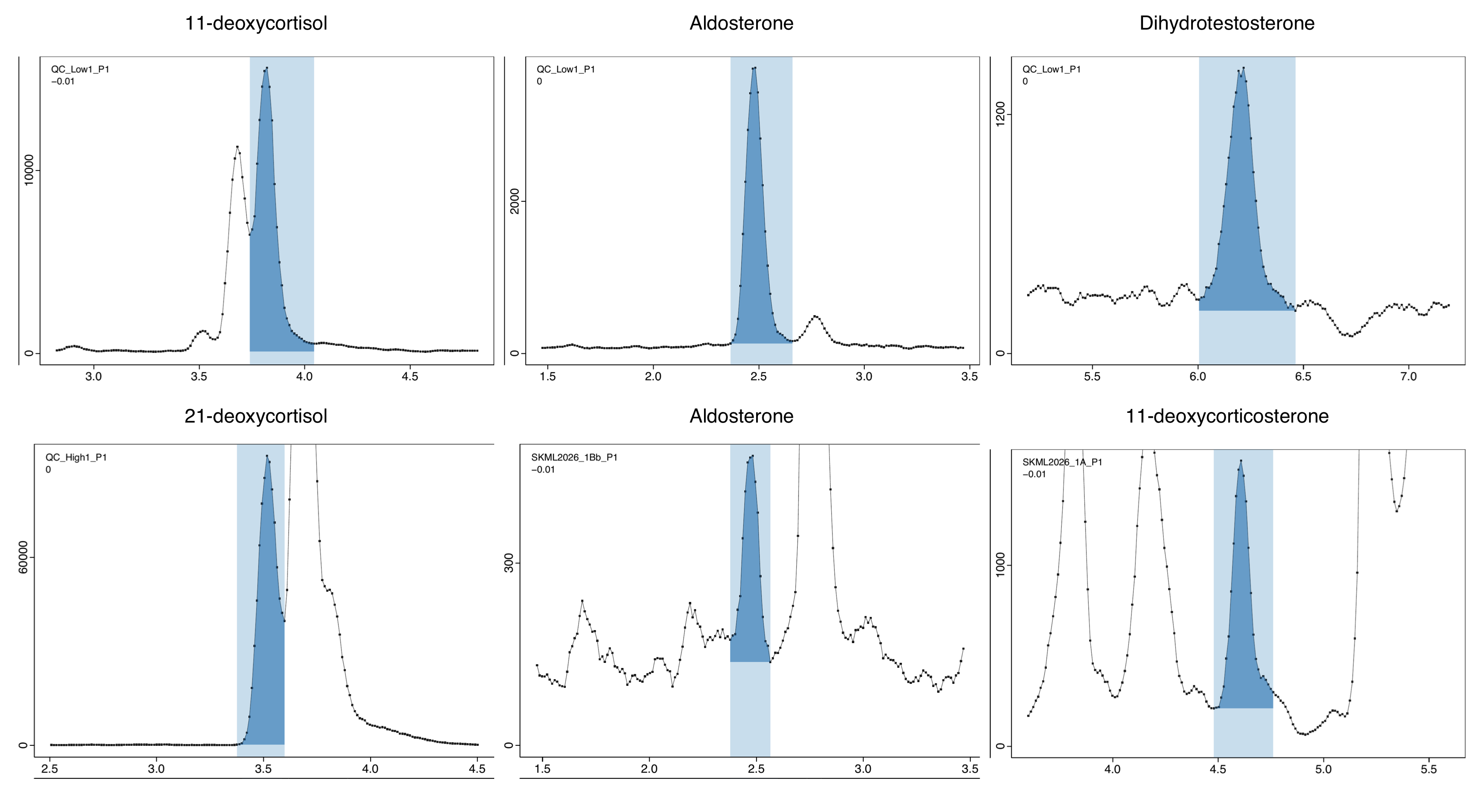
QC_Low1): 11-deoxycortisol, Aldosterone, Dihydrotestosterone. Bottom row — three demanding cases, each with a co-eluting neighbour: 21-deoxycortisol in the high QC (QC_High1, large neighbour to the right), Aldosterone in EQA sample SKML2026_1Bb (neighbour to the right, peak at 0.3 × Cal A) and 11-deoxycorticosterone in SKML2026_1A.
3 Import
Data and corresponding metadata are loaded into a fresh MRMhubExperiment. import_data_mrmhub() reads the INTEGRATOR long-format peak areas for the assay batch (blanks, calibrators, QCs and EQA samples). import_metadata_msorganiser() then attaches mrmhub’s metadata workbook template, which defines the analytes, their internal standards, the sample annotations, and the calibration/target concentrations.
mexp <- MRMhubExperiment(title = "Dataset 4 — Steroid Assay")
mexp <- import_data_mrmhub(
mexp,
path = "./data/dataset-4/Dataset4_MRMhub-INTEGRATOR_ASSAY.csv",
import_metadata = TRUE
)✔ Imported 20 analyses with 64 features.ℹ feature_area selected as default feature intensity. Modify with `set_intensity_var()`.✔ Analysis metadata associated with 20 analyses.✔ Feature metadata associated with 64 features.mexp <- import_metadata_msorganiser(
mexp,
path = "./data/dataset-4/Dataset4_Metadata.xlsx",
excl_unmatched_analyses = TRUE, ignore_warnings = TRUE
)Found no errors, 2 warnings, and 2 notes in the metadata.
----------------------------------------------------------------
Type Table Column Issue Count
1 W* Analyses analysis_id Analyses not in analysis data 12
2 W* Features feature_id Feature(s) without metadata 34
3 N Analyses sample_id Not defined for all analyses 16
4 N Features analyte_id Not defined for all features 15
----------------------------------------------------------------
E = Error, W = Warning, W* = Suppressed Warning, N = Note
----------------------------------------------------------------✔ Analysis metadata associated with 20 analyses.✔ Feature metadata associated with 30 features.✔ Internal Standard metadata associated with 15 ISTDs.✔ QC concentration metadata associated with 13 samples and 15 analytes4 Calibration
normalize_by_istd() divides each analyte’s peak area by that of its authentic stable-isotope-labelled internal standard. calc_calibration_results() then fits one external calibration curve per analyte from the Cal A–Cal F levels. A quadratic model with 1/x weighting was used as the regression model for all analytes.
mexp <- normalize_by_istd(mexp)✔ 15 features normalized with 15 ISTDs in 20 analyses.mexp <- calc_calibration_results(
mexp,
fit_overwrite = TRUE,
fit_model = "quadratic",
fit_weighting = "1/x"
)✔ Calibration curve fits calculated for all 15 quantifier features. Average r²: 0.9978.# Curves are solid between the lowest and highest calibrator, and dotted where
# extrapolated beyond that calibrator range (automatic; not a range restriction).
plot_calibrationcurves(
mexp,
fit_overwrite = FALSE,
fit_model = "quadratic",
fit_weighting = "1/x",
include_istd = FALSE,
include_qualifier = FALSE,
rows_page = 4,
cols_page = 4,
show_progress = FALSE
)✔ Calibration curve fits calculated for all 15 quantifier features. Average r²: 0.9978.✔ Done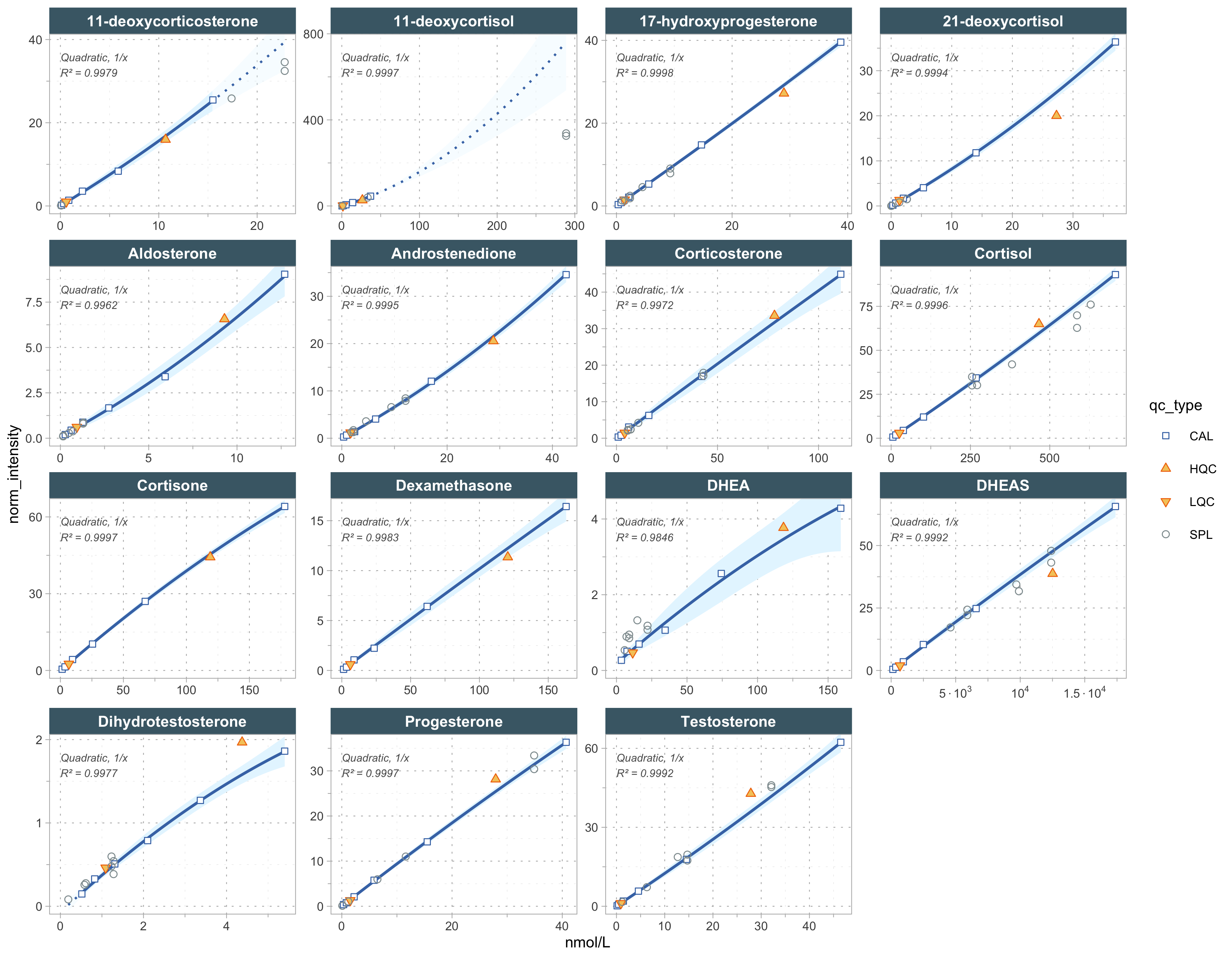
The metrics below summarize each quantifier’s fit: R² (linearity/goodness of fit) and the estimated LOD and LOQ (limits of detection and quantification, in the calibrated concentration unit).
get_calibration_metrics(mexp, include_qualifier = FALSE, summary_table = TRUE)5 Quantification
quantify_by_calibration() inverts each fitted curve to convert the normalized intensities into absolute concentrations. Only quantifier transitions are used (include_qualifier = FALSE), and ignore_failed_calibration = TRUE skips any analyte whose curve failed to fit rather than aborting the whole run, so a single problematic analyte does not block the report. fit_model and fit_weighting can also be set per analyte in the feature metadata table.
mexp <- quantify_by_calibration(
mexp,
fit_overwrite = FALSE,
include_qualifier = FALSE,
ignore_failed_calibration = TRUE,
fit_model = "quadratic",
fit_weighting = "1/x"
)✔ Calibration curve fits calculated for all 15 quantifier features. Average r²: 0.9978.ℹ 77 concentration values fall outside the calibrated range (retained, flagged in feature_conc_out_of_range).✔ Concentrations calculated for 15 features in 20 analyses.✔ Concentrations are given in nmol/L.6 QC bias & variability
The final check compares the measured QC concentrations against their known targets. get_qc_bias_variability() reports, per analyte and QC level, the mean measured concentration, the bias (percent deviation from target — accuracy) and the intra-batch %CV (precision), plus a flag for values falling outside the calibrated range. Restricting to LQC/HQC keeps the table to the samples that have assigned target concentrations.
get_qc_bias_variability(mexp, qc_types = c("LQC", "HQC"), summary_table = TRUE)7 Export
The results are written in three complementary formats, each for a different use.
7.1 Flat concentration table (CSV)
save_dataset_csv() writes a single flat table of concentrations, one row per sample — the simplest format to open in Excel or read into any tool. add_qctype keeps the QC-type column so calibrators, QCs and samples stay distinguishable.
save_dataset_csv(
mexp,
path = file.path("output", "Dataset4_report_conc.csv"),
variable = "conc",
add_qctype = TRUE
)✔ Concentration values for 20 analyses and 15 features have been exported to 'output/Dataset4_report_conc.csv'.7.2 Multi-sheet report (XLSX)
save_report_xlsx() writes a multi-sheet workbook bundling the concentrations and the supporting context (calibration metrics, feature QC metrics, raw/normalized intensities, sample and feature metadata) in one file for sharing and archiving.
save_report_xlsx(
mexp,
path = file.path("output", "Dataset4_report.xlsx"),
filtered_variable = "conc",
overwrite = TRUE
)✔ The data processing report of experiment 'Dataset 4 — Steroid Assay' has been saved to output/Dataset4_report.xlsx.7.3 Complete dataset object (RDS)
save_dataset_rds() writes the entire MRMhubExperiment — imported data, metadata, calibration fits and quantification results — to a single .rds, so that everything needed to reproduce, extend or audit the analysis is contained in one file. An rlang::hash() fingerprint is embedded, allowing the file’s integrity to be verified when it is read back. The file is a plain R object and can therefore be opened in any R session with readRDS(), even without mrmhub installed; with mrmhub loaded, the object’s methods are available and the analysis can be continued from where this report ends.
save_dataset_rds(mexp, "output/Dataset4_report_mexp.rds")✔ MRMhubExperiment saved to output/Dataset4_report_mexp.rds.Content fingerprint: "015a1a3574e523721bb58f35ce6127fe"Reload it in a fresh R session with read_dataset_rds(), which verifies the embedded integrity hash on read (load mrmhub first so the object’s methods are available):
library(mrmhub)
mexp <- read_dataset_rds("output/Dataset4_report_mexp.rds")8 References
Jansen, Rob, Nuthar Jassam, Annette Thomas, et al. 2014. “A Category 1 EQA Scheme for Comparison of Laboratory Performance and Method Performance: An International Pilot Study in the Framework of the Calibration 2000 Project.” Clinica Chimica Acta 432 (May): 90–98. https://doi.org/10.1016/j.cca.2013.11.003.