Correcting isotopic interference
Source:vignettes/articles/tutorial-11-interference-correction.Rmd
tutorial-11-interference-correction.RmdMRMhub corrects isotopic (M+2) overlap between co-eluting same-class
lipids with LICAR (Gao et al. 2021). It
runs on the raw feature_intensity and, unlike drift and
batch correction, applies to every sample. This tutorial annotates the
pattern, derives the overlaps, inspects them, and subtracts them; the Isotopic interference
correction manual covers the concepts, the derivation levels, and
the full mrm_pattern list.
1. Annotate the MRM pattern
Automatic derivation needs one hand-added column,
mrm_pattern, on the Features metadata sheet;
it names the lipid class and the product-ion type (the manual lists
every label and shows the column in context). The valid labels for a
product-ion type are returned by
licar_pattern_choices():
library(mrmhub)
licar_pattern_choices("Head Group")MRMhub validates the label on import. Load a processed object to continue:
mexp <- readRDS("results/mexp_processed.rds")2. Derive the interference relationships
calc_isotopic_interferences()
discovers the M+2 overlaps and stores them in
annot_interferences. Set level = "MRM" for
class-based LC-MRM; "MS1" is only for genuine full-scan
data, not a fallback when a product m/z is missing (see the
manual).
# Class-based LC-MRM: fragment-level M+2 (needs precursor + product m/z).
mexp <- calc_isotopic_interferences(mexp, level = "MRM")
# Genuine MS1 / full-scan: whole-molecule M+2 (needs precursor m/z only).
# mexp <- calc_isotopic_interferences(mexp, level = "MS1")Derivation is deterministic; raise min_contribution to
drop negligible pairs.
3. Inspect the derived relationships
Before subtracting, review the derived overlaps. Each row of
annot_interferences is one overlap, described by its
overlap_type and source columns (defined in
the manual):
mexp@annot_interferencessummarize_interferences()
rolls this up per feature: how many are affected, split by source and
overlap type, with the contribution-factor range.
summarize_interferences(mexp)4. Apply the correction
correct_isotopic_interferences()
subtracts all derived overlaps from the raw intensities in one pass (its
reference page gives the formula and zero-clamping).
mexp <- correct_isotopic_interferences(mexp)Apply it before ISTD normalisation, drift, and batch correction;
running it later resets those steps (with a warning). Uncorrected values
are kept in feature_intensity_orig. Overlaps you annotate
yourself (in-source fragments, co-eluting isobars) are applied with correct_custom_interferences().
5. Correct a single pair manually
For a one-off correction, or to check a factor before trusting the
automatic derivation, use correct_interference_manual(),
where variable is the dataset column:
mexp <- correct_interference_manual(
mexp,
variable = "feature_intensity",
feature = "PC 32:0",
interfering_feature = "SM 36:1 M+3",
interference_contribution = 0.0107,
neg_to_na = FALSE,
updated_feature_id = NA)updated_feature_id renames the corrected feature so the
raw and corrected channels can coexist.
6. Verify the correction
Compare a corrected feature’s intensities before and after; in blanks
(SBLK/PBLK) the residual signal should
approach zero, and a non-zero blank median points to an underestimated
factor.
d <- get_analyticaldata(mexp, annotated = TRUE) |>
dplyr::filter(feature_id == "PC 32:0") |>
dplyr::select(
analysis_id, qc_type,
intensity_before = feature_intensity_orig,
intensity_after = feature_intensity) |>
dplyr::mutate(
pct_change =
100 * (intensity_after - intensity_before) / intensity_before)
summary(d$pct_change)plot_qc_interference_impact()
shows how many features were corrected at each magnitude (percent of
signal removed):
plot_qc_interference_impact(mexp, qc_types = "SPL")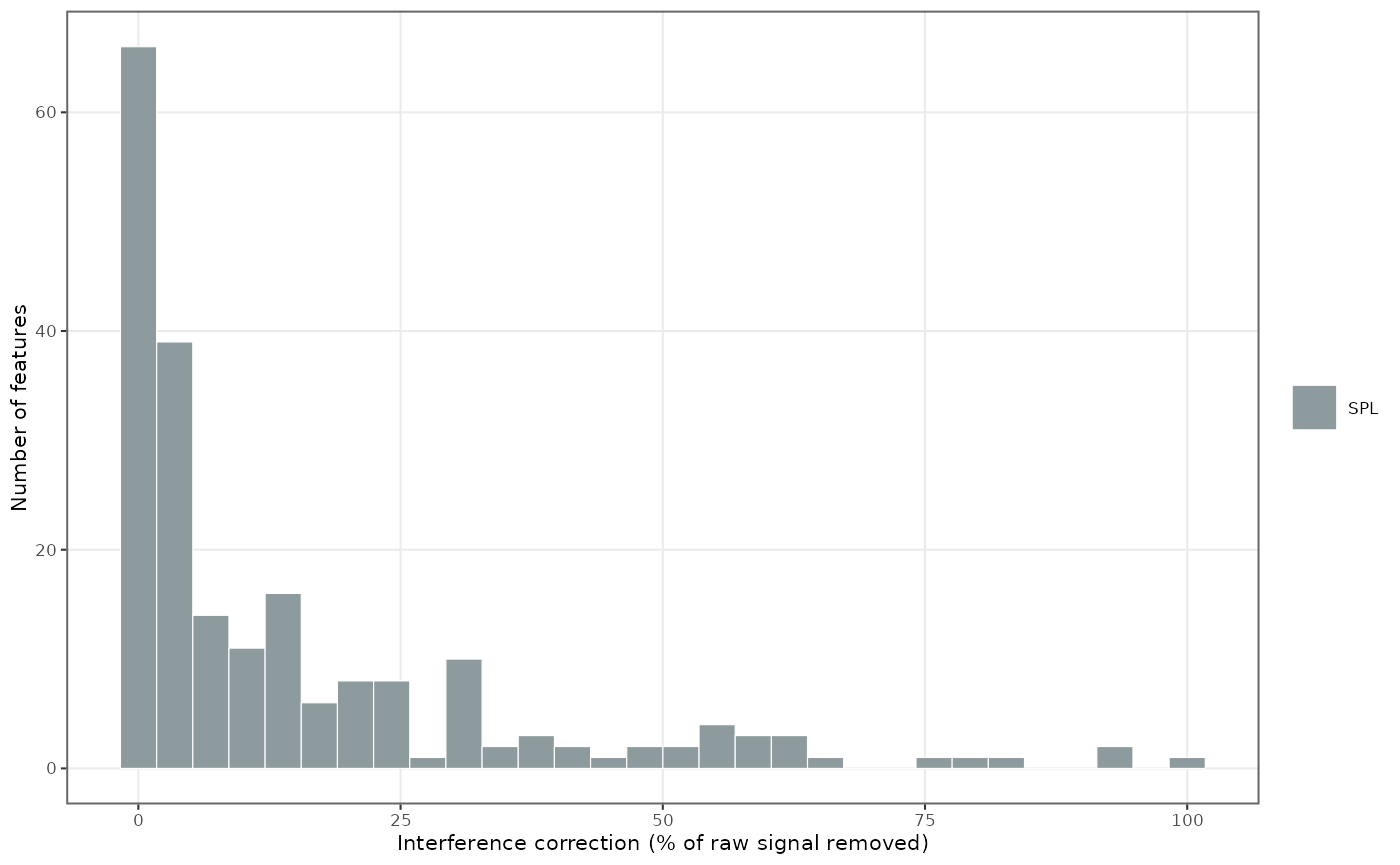
Figure 1. How many features were corrected at each magnitude (percent of signal removed) in the study samples.
plot_interference_correction()
plots each feature’s residual signal as a percent of its uncorrected
value, split by QC type. min_correction_pct restricts it to
strongly affected features (here ≥ 40 % removed),
sort_by_effect ranks by magnitude, and top_n
keeps the largest:
plot_interference_correction(
mexp,
qc_types = "SPL",
min_correction_pct = 40,
sort_by_effect = "desc",
point_size = 1.5, point_alpha = 0.8)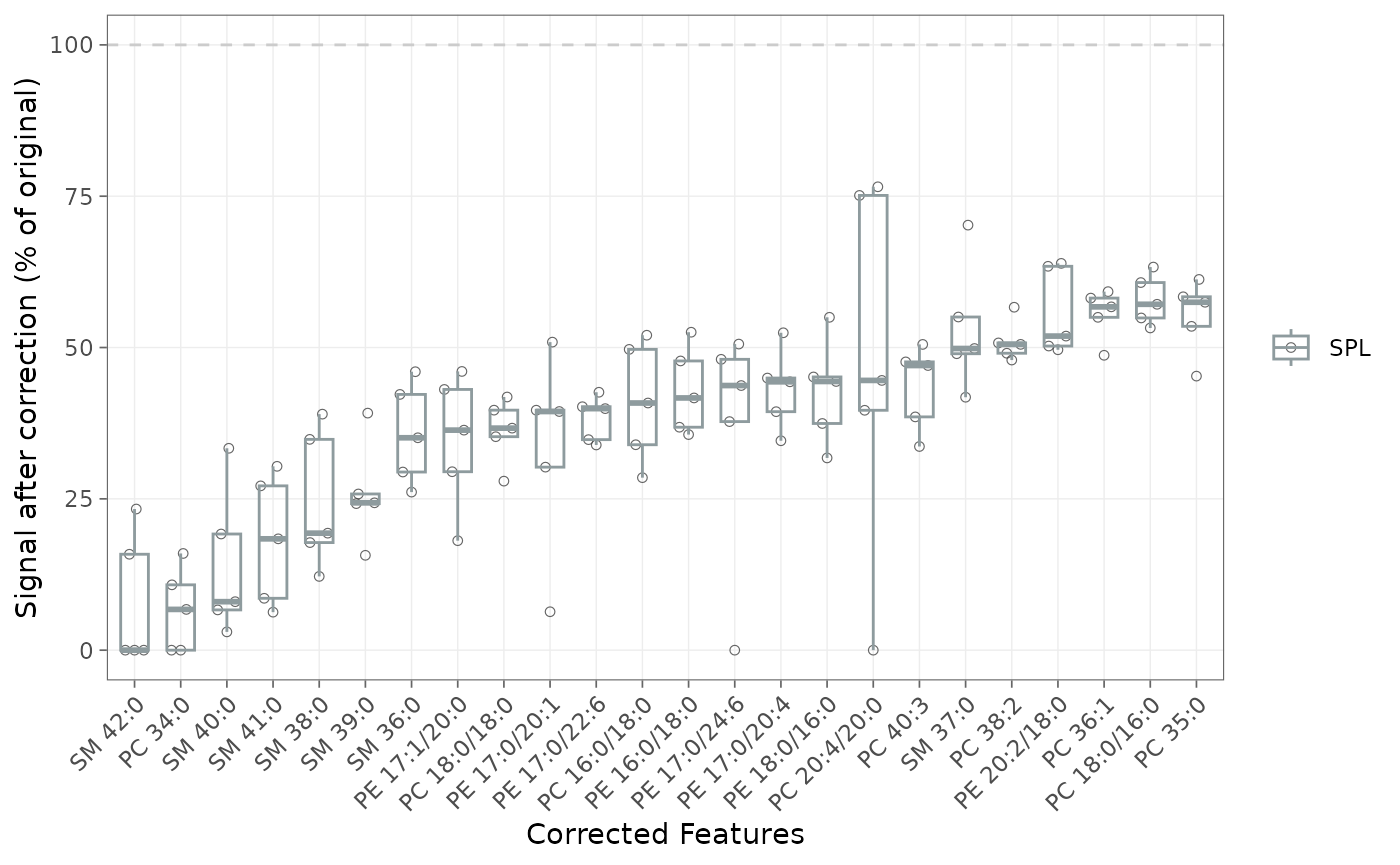
Figure 2. Per-feature residual signal after correction, as a percent of the uncorrected value, for features with at least 40% removed, split by QC type.
Co-elution filtering (experimental)
check_coelution is experimental and off
by default. Verify the pairs it keeps and drops on your own data before
relying on it.
The correction subtracts the full source area, so it is valid only
where the source and target peaks co-elute and/or are co-integrated. The
experimental check_coelution gate keeps only the
m/z-matched edges that pass this test and drops
chromatographically resolved pairs (see the manual):
mexp <- calc_isotopic_interferences(
mexp, level = "MRM", check_coelution = TRUE)It is off by default while the gate is validated, and reports how many pairs it drops.
Next steps
- Isotopic interference correction (manual): concepts, derivation levels, and the full label list
- Drift and batch correction: apply after interference correction
- Lipidomics workflow: the full processing pipeline
-
The
MRMhubExperiment data object: how
_origpreserves the raw intensities